Your gift is 100% tax deductible.
Retinoblastoma
Retinoblastoma is a rare cancer of the eye. Most retinoblastoma is diagnosed in young children. If your child or someone you are close to has retinoblastoma, knowing what to expect may be helpful. Here you can find out more about retinoblastoma, including risk factors, symptoms, and how tumors are found and treated.
About retinoblastoma
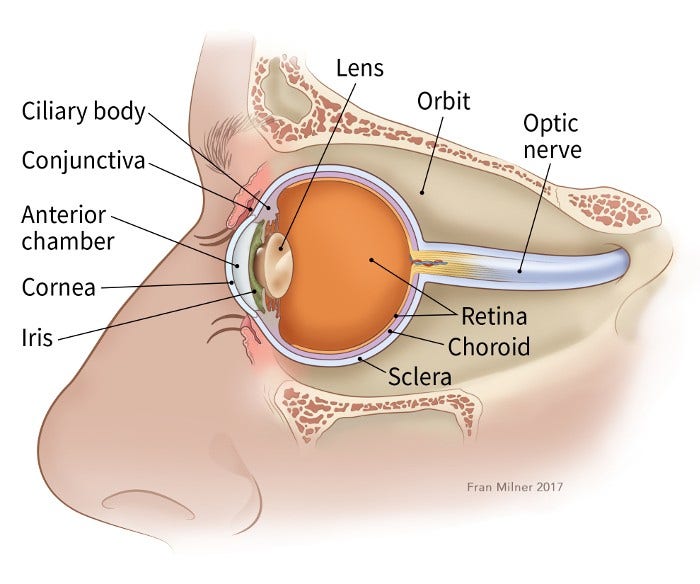
Retinoblastoma is a cancer of young children that starts in the retina, which is a part of the eye.
The eyes start to develop well before birth. During the early stages of development, the eyes have cells called retinoblasts, which multiply to make new cells that fill the retina. At a certain point, these cells stop multiplying and become mature retinal cells.
Understanding the retina
The retina is the inner layer of cells in the back of the eye. It is made up of special nerve cells that are sensitive to light. These light-sensing cells are connected to the brain by the optic nerve, which runs out of the back of the eyeball. The pattern of light (image) that reaches the retina is sent through the optic nerve to an area of the brain called the visual cortex, allowing us to see.
Rarely, instead of maturing, some retinoblasts continue to grow out of control, forming retinoblastoma. Retinoblastoma tumors can be:
- Unilateral, affecting one eye
- Bilateral, affecting both eyes
- Trilateral, a condition where a child with retinoblastoma also develops a brain tumor, often near the middle of the brain in an area called the pineal gland
Retinoblastoma often starts because of a change (mutation) in a gene called RB1. About half of children with retinoblastoma have the heritable type, meaning the RB1 gene is abnormal in cells throughout the body, not just in the tumor. This is important to know because these children have a higher chance of other types of cancer later in life.
Quick guides
- Written by
- References

Developed by the American Cancer Society medical and editorial content team with medical review and contribution by the American Society of Clinical Oncology (ASCO).
Berry JL. Retinoblastoma: Clinical presentation, evaluation, and diagnosis. UpToDate. Accessed at www.uptodate.com/contents/retinoblastoma-clinical-presentation-evaluation-and-diagnosis on July 2, 2025.
Leahey AM, Gombos DS, Chevez-Barrios P. Chapter 32: Retinoblastoma. In: Blaney SM, Adamson PC, Helman LJ, eds. Pizzo and Poplack’s Pediatric Oncology. 8th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2021.
National Cancer Institute. Retinoblastoma Treatment (PDQ®). 2025. Accessed at www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq on July 2, 2025.
Last Revised: September 12, 2025
American Cancer Society medical information is copyrighted material. For reprint requests, please see our Content Usage Policy.
This information is possible thanks to people like you.
We depend on donations to keep our cancer information available for the people who need it most.